📗 -> 05/13/25: NPB173-L11
🎤 Vocab
❗ Unit and Larger Context
Midterm 2 Review Lecture
- Overview of major concepts
- Review of more challenging material
- Review of submitted exam sample questions
✒️ -> Scratch Notes
Review up until now
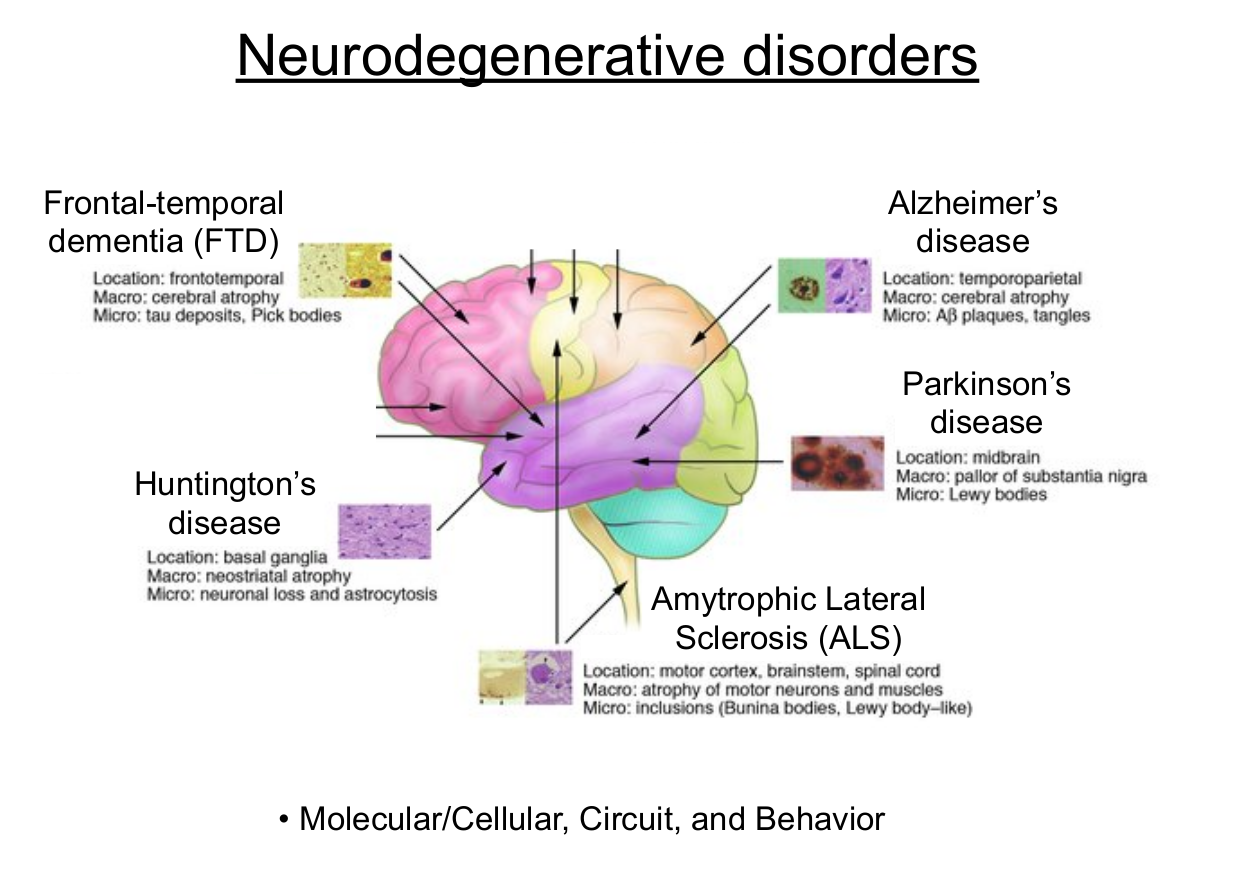
Lecture 8: Huntington’s Disease
- Common features across neurodegenerative diseases
- Selective vulnerability
- Different celltypes for different diseases
- Comes about from some abnormality that preceded vulnerability
- Progresses slowly over time (course of lifetime even)
- Cell death started to occur later in life
- With death, function start to deteriorate and we see symptoms increasing over time
- Selective vulnerability
- Neurodegenerative diseases across spatial scales
- Understand how the diseases come about across scales
- Understand each disease
- Mo- Wdo we even know??lecular -> cellular -> circuits -> Nervous System -> Function
- Inheritance of neurodegenerative diseases
- One of the most understood, understand the best
- Singular cause of disease
- Clear genetic cause and pattern of inheritance (especially for Huntington’s)
- Autosomal dominance and full penetrance in Huntington’s, other cases more fuzzy if even inherited
- Mendelian inheritance
- Understand and do calculations to make predictions based on genetics of parents (e.g. Punett Square)
- Central dogma of molecular biology
- Understand DNA replication, what problems can happen, how mutations can occur
- Replication -> protein expression -> Transcription -> Translation
- 3 consecutive nucleotide pairs in a codone, correspond to amino acids, coded by exons of a gene, critical components of a primary structure of a protein
- Signs and symptoms of Huntington’s
- Symptoms (1st person):
- Huntington’s Corea, earliest salient symptom
- Flailing movements -> Rigidity -> Cognitive impairment over time
- Signs(3rd person):
- Genetics, genetic tests. Not a symptom, cannot introspect
- Symptoms (1st person):
- Pre-manifest and manifest stages of progression
- Pre-manifest - Before diagnosis
- Presymptomatic - Before there are symptoms
- Prodromal - Symptoms are potentially apparent but not severe enough for diagnosis.
- Transition phase
- Manifest - After a diagnosis has been reached. Here, mostly talking about symptoms, ignoring genetic tests.
- Pre-manifest - Before diagnosis
- Huntingtin genetic mutation and relation to Huntington’s
- Gene discovered after the disease, named after the disease name (not a typo lmao). -in is traditional for gene and protein naming.
- Key aspect of mutation is number of CAG (glutamine) repeats.
- Fewer than 36 repeats (<36) they will not develop Huntington’s
- 40 or more (>40) they will develop Huntington’s, Full penetrance
- 36-39 reduced pentrance for Huntington’s.
- If you have 35, you yourself will not get Huntington’s but your children will be at risk.
- On average, comes out to Mendelian inheritance (if only 1 parent has it, 50% chance child will have it)
- Abnormal Huntingtin protein
- Corresponds to CAG repeats as described above
- Gain of functions vs. loss of function
- Problems can be caused through two classifications
- Lose one of its normal functions (loss of function)
- Protein does something new and different (gain of function)
- Good, bad, or even complex (could be helpful in some ways and harmful in others)
- Not divided into good or bad, but by the funtion it gains/loses
- Most abnormalities discussed are by and large more gain of function than loss of function
- Problems can be caused through two classifications
- Neural inclusions and protein aggregates
- One pattern seen is that protein abnormalities across neurodegenerative diseases is that they will form protein aggeregates. Sloooowly across lifetimes.
- As they form, they stabilize such that proteins that would be normally broken down are not, lingering longer.
- When aggergate large enough, readilly visible
- Huntington’s: Huntington’s Neural Inclusions / Neural Inclusions
- Lewy Body inclusions
- Neurofibular tangles
- Amyloid Plaques
- When aggergate large enough, readilly visible
- Inclusions are not solely protein, but have a large amount of abnormal proteins associated with disorder
- Inclusions are not the toxic components, not directly the problem
- Earlier in the cascade there was a problem
- Acquired gain of function by having the aggregate that causes a problem in the cell
- Evidence for gain doesn’t mean that loss of function is not the case. We just know there is gain of function.
- Very possible that loss of function is also happening in Huntington’s
- Neural circuits affected by Huntington’s
- Striatal circuits in particular here
- Connect circuits to signs and symptoms
- Degeneration of indirect pathway neurons in the striatum is thought to be responsible for flailing movements in corea
- Degeneration of direct thought to be some of the motor control and rigidity problems
Additionally, try to map out the “full story” from different levels of analysis. This is best known in Huntington’s for example:
- Autosomal dominant allele for Huntingtin gene is inherited with abnormally large number of CAG repeats.
- This causes expression of the abnormal Huntingtin protein with long PolyQ tract
- Fragments of abnormal protein lead to neuronal degeneration
- Not the full protein, deduce that protein undergoes cleavage which leads to toxic component/aggregate which causes problems and leads to inclusions
- Degeneration disproportionately affects circuits vulnerable to the abnormality in the striatum.
- Explained above in “Neural circuits affected” bullet point
- Affected circuits in striatum play a critical role in motor control.
- Motor control impairments manifest at a behavioral level
Lecture 9: Parkinson’s Disease
Thematically similar to Huntington’s, targeting similar circuits.
- Basic concepts related to neurotransmitter synthesis
- Dopiminergic system is affected dramatically, understand what’s going on
- Huntington’s too, striatal neurons using GABA affected heavily.
- Contrast between Dopamine as a neuromodulator and GABA as ionotropic neurotransmitter
- Basic concepts related to neurotransmitter clearance
- Consider whole life cycle of neurotransmitters
- Diffusion / Degradation / Reuptake
- Specifics of the dopamine life cycle
- Do know basic features for all neurotransmitters
- Common precursors, like amino acids. Prevalent across cells
- What really dictates synthesis is the protein enzymes. THIS is the rate limiter
- Might be multiple steps, and might have multiple transmitters in the same pathways
- Recapture can occur through neurons (presynaptic often) or glial (astrocytes)
- Astrocytic reuptake of glutamate is important excitotoxicity
- Do know basic features for all neurotransmitters
- Basic concepts related to ascending neuromodulatory systems
- Dopamine is part of an ascending neuromodulatory systems
- Basic features of neuromodulatory systems:
- Generally: subcortical sources of cell bodies that project distinctly for different systems but diffusely to many other regions
- Characterized by their neurotransmitter
- Neurotransmitter has effects through metabotrophic receptors, so not directly on ion channels but through second messenger cascades
- Effects are slower but longer lasting
- Specifics of basal ganglia circuits as discussed in class
- Signs and symptoms of parkinson’s disease
- Signs:
- Symptoms:
- Bradykenisia, slowness of movement.
- Characteristic gait, characteristic turn
- Motor system rigidity and tremor. General impairment of motor control from impairment to basal ganglia
- Familial vs. sporadic diseases and relation to age of onset
- Defined by patterns of inheritance instead of raw genetics. Related of course.
- Familial - If afflicted individuals family members had the disease at a rate higher than general population. “it looks like it’s inherited”. Not equivalent to genetics, might be recessive in family and not expressed
- Sporadic - All other cases. Not from having family members having it.
- Onset:
- familial generally expresses earlier than sporadic
- If its familial, we can likely infer a genetic component
- Sporadic does not rule out genetic component, but suggests no singular genetic cause with high penetrance
- Genetic factors affecting neurodegenerative disorders
- Can have monogenetic factors (like Huntingtons) but can also have polygenetic factors (combined effects), or genes that only lead to higher/lower risk.
- Need to mix them all to determine whether disorder will be developed
- Can have monogenetic factors (like Huntingtons) but can also have polygenetic factors (combined effects), or genes that only lead to higher/lower risk.
- Multiple initiating causes with a final common pathway of dysfunction
- The ‘canyon’ analogy, of multiple paths to the common pathway.
- Not as applicable for Huntington’s, that has one straightforwards cause and path
- Parkinson’s is a clear example. Final common path is degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNC)
- Cases where people have autosomal dominant mutation that leads to it, and cases where people don’t have that, family history, or other clear genetic factors (sporadic)
- The ‘canyon’ analogy, of multiple paths to the common pathway.
- SNCA gene, alpha-synuclein protein, and Lewy bodies
- Molecular abnormalities:
- Alpha synuclein
- SNCA gene corresponds to alpha synuclein?
- People develop parkinson’s with SNCA gene
- Almost everyone with Parkinson’s involves alpha-synuclein protein and aggregates (Lewy bodies)
- Parkinsonian: Anything that results in Parkinson’s-like symptoms due to degeneration of dopaminergic neurons
- A stroke that takes out SNC neurons
- A drug that kills dopaminergic neurons
- Lewy bodies also associated with disorders other than Parkinson’s
- Synuclein kind of implied by SNCA, helpful?
- Molecular abnormalities:
- Circuit mechanisms of Parkinson’s: rate model and pattern hypothesis
- Braak staging and progression of degeneration in Parkinson’s
- Orderliness of progression
- Not the same for different proteins (differences between tau and amyloid), but general orderliness
- Possible treatments for Parkinson’s
- Think about different scales:
- LDOPA, boost dopamine in cells that have not degenerated
- Does not prevent degeneration itself
- Purely treats symptoms, not a cure, but is beneficial
- In the past was treated by lesions to help overcome bradykinesia
- DBS
Dopamine Life Cycle:
Know synaptic cleft life cycle
Synthesis:
- Tyrosine
-
Tyrosine hydroxylase
- THIS IS THE RATE LIMITING STEP.
- Shit tons of tyrosine, limited by enzyme
- Dihydroxy-phenylalamine (DOPA)
-
Amino acid decarboxylase
- Dopamine
Degradation:
Dopamine can be degraded into:
- MAO (monoamine oxidase), aidehyde dehydrogenase
- COMT (catechol-O-methyl-transferase)
Ends up as DOPAC or 3-MT
Further degraded into homovanillic acid (HVA)
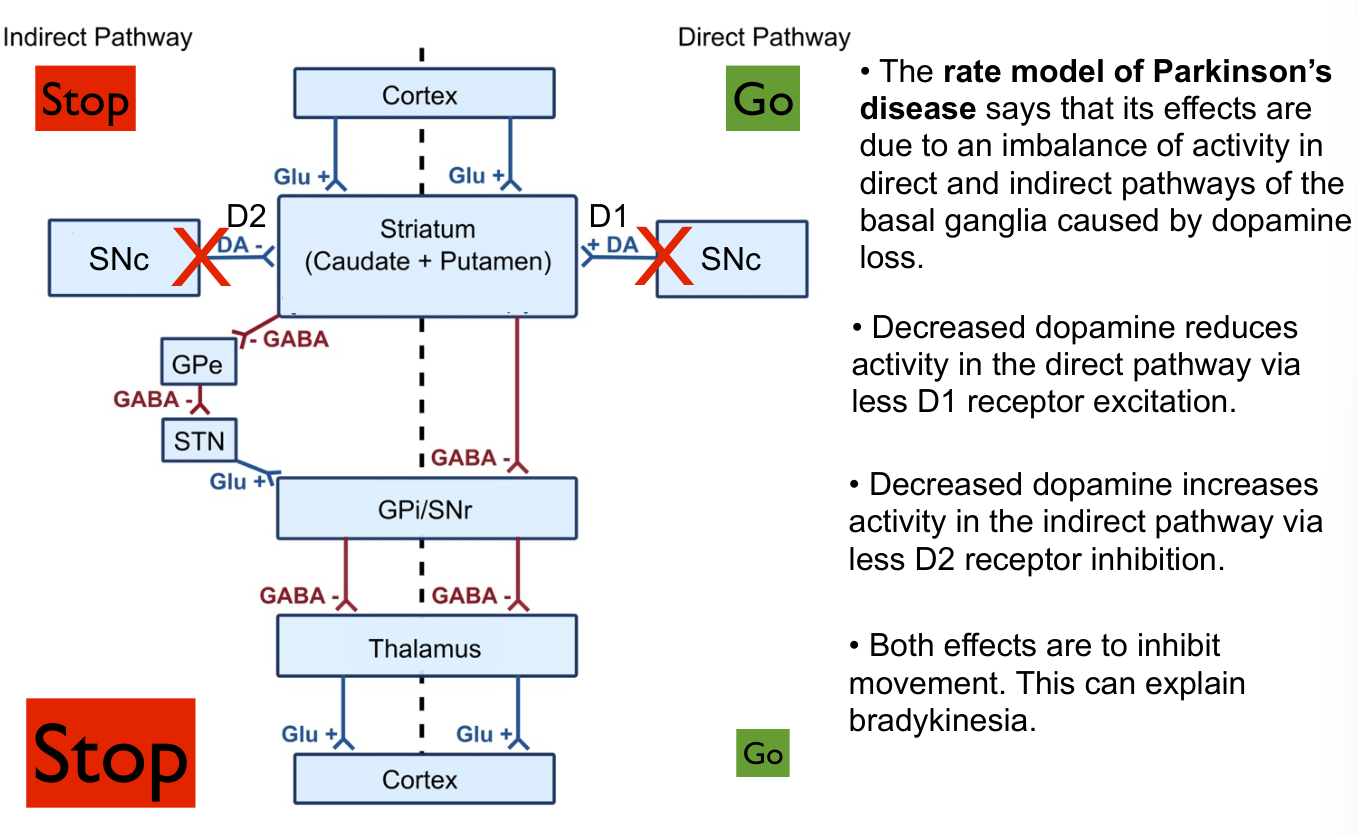
- Indirect pathway on the left
- D2 receptors, generally inhibited with dopamine
- Direct pathway on the right
- D1 receptors, generally excitation with dopamine
Dopamine leads to more go, less stop.
- D1 receptors, generally excitation with dopamine
- Overall dopamine leads to more excitation
Hypothesis: - Parkinson’s shifts the balance towards stop (rate hypothesis)
- Pattern hypothesis - Parkinson’s activity is more bursty than normal. This is theorized to play a role in problems
- They are not mutually exclusive, both likely play a role.
Striatum is a part of both, distinct
SNc - Substancia Nigra Compacta
STN - Sub thamalamic nucleus
GPe - Global paladus external. Indirect.
GPi - Global pallidus internal. Direct
Most connections are inhibitory!!
- Everything in basal ganglia circuit are inhibitory aside from cortex or things with thalamus in the name
- Glutamate: Thalamus, STN, Cortex
- Dopamine: SNc
Lecture 10: Alzheimer’s Disease
One of the ‘big ones’ from an overall impact perspective.
Some of the most intricate details are known about these as well, very well researched.
-
Proteostatic control beyond the central dogma of molecular biology
- Proteins that come back and regulate the processes
- Transcription factors
- Not just a linear flow with no feedback
- DIfferent subsets of genes can be splices to different ends
- Proteins that come back and regulate the processes
-
Post-translational modifications and protein targeting
- Basic categories of reversible and irreversible moditifications
- Some specifics related to alzheimers
- Cleavage and order of cleavage
- Not reversible
- Tau phosphorylation a critical component for buildup of tau aggregates and dissociation of tau from the microtubules
- Phosphorylation IS reversible, could be targeted for treatment
- Cleavage and order of cleavage
-
Microtubule-mediated axonal transport
- Neurons have very long axons, instructions for protein expression are in the nucleus in the DNA, which can be far from where DNA needs to act
- Neurons have very difficult protein targeting
- It is mediated by microtubules
- Energetically demanding
- If disrupted, will lead to problems/disfunctions at axonal terminals
- Selected vulnerabilities:
- Might target longer axons vs shorter
-
Signs and symptoms of Alzheimer’s disease
-
Signs:
- Symptoms:
- Cogntive decline
- Memory
- MCI (mild cognitive impairment) - Impairment worse than typical aging, but not so bad it interferes with caring for oneself. Patients usually aware of this, and its abnormal.
- Dementia: Severe cognitive impairment and memory loss
- Cogntive decline
-
-
Amyloid plaques and neurofibrillary tangles
- At the level of proteins, two salient features:
- Amyloid plaques - Extracellular aggregates (outside neurons)
- Neurofibrillary tangles - Fibrous-like aggregates that extend the entire length of axons inside of neurons. Include tau proteins
- At the level of proteins, two salient features:
-
Mechanisms involved in A
-42 formation - Most attention for neuron, but controversial around clinical trials.
- Years and years of unsuccesffuly trials, recently a few more succesffuly ones. Aiming to target earlier
- We know that plaques involve a protein fragment (Abeta, Abeta-40 and Abeta-42)
- Most attention for neuron, but controversial around clinical trials.
-
Genetic mutations that affet A
-42 levels and how they work - Mutations:
- Directly on precursor protein (protein that Abeta40/42 are part of)
- and protein that are close to the cleavage sites
- What could increase/decrease production of 40/42?
- Of the mutation that has highest penetrance it is about the proteins involved in the cleavage: presynillins. Discovered in the search for alzheimers.
- By changing probability of cleavage, can affect ab40/42 and ab overall
- Presynillin mutations have earlier onset of alzheimers
- Early onset alzheimers
- Large fraction of cases have identifiable genetic mutations
- Mutations:
-
Tau protein and neurofibrillary tangles
- How important is tau vs amyloid?
- Tau is a protein that is also expressed at high levels within neurons, stabilizes microtubules
-
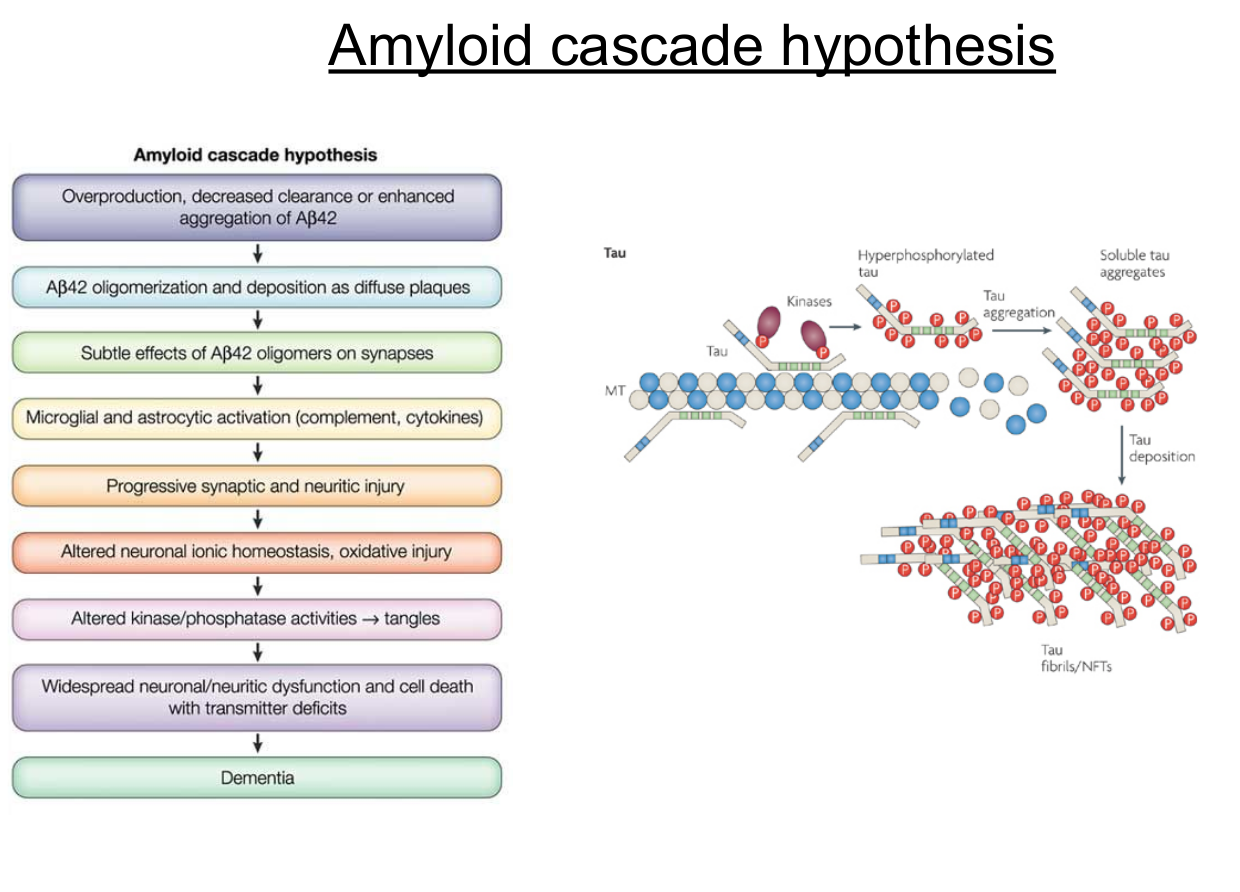
Amyloid cascade hypothesis
-
Neurodegeneration in Alzheimer’s and systems affected
- At the molecular scale, we can talk about protein abnormalities
- Connect to circuit scale to selective vulnerabilities
- HPC, general cortex, temporal cortex especially
- HPC: formation of new LTM
- People seem to not form long term memories in MCI
- Degeneration in frontal as disease progresses to
-
Cholinergic hypothesis for Alezheimer’s
- Also have early degeneration in neuron’s that use acetylcholine as their neurotransmitter
- Part of cholinergic ascending neuromodulatory system
- used to be a hot theory about alzheimers
- By and large, not considered analogous to parkinson’s
- Also have early degeneration in neuron’s that use acetylcholine as their neurotransmitter
-
Other types of dementia
- Alzheimer’s not the only dementia
- Frontal temporal dementia
- Vascular dementia - overall cognitive decline without being caused by alzheimers
- Same circuits affected, same deficits
- Same circuits affected, same deficits
-
People arguing that decreased clearance is an early factor in alzheimers
- People argue clearance >> production
-
APOE - protein involved in clearance of many molecules including A-beta. A major risk factor depending on APOE alleles
-
The idea is that some step in the cascade causes hyperphosphorylation of tau, which causes to it dissacoiation from microtubules, a breakdown of the microtubules, and cell death
-
Models that include only tau, part of the final pathway
-
Two molecular mechanisms: amyloid and tau
- Amyloid: amyloid precursor fragment is embedded in the membrane, the fragment partially embedded in membrane
- 3 main cleavage sites: you get ab40/42 when you:
- Get first cut at the beta site (but not the alpha, that will interrupt, splitting it)
- Then get cleavage in either the gamma40 or gamma42 site
- 40/42nd amino acid
- 42 thought to be more toxic
- likely doing good things, but over time building up and doing bad things as well
- 3 main cleavage sites: you get ab40/42 when you:
- Tau: Neurofibullary tangles. Tau help to stabilize microtubules with multiple phosphorilation sites.
- These sites are regulatory mechanisms for controlling association of stabilizing proteins on the microtubules, potentially could help to break down microtubules (could be useful in Wallerian degeneration!)
- When they got hyperphosphorylated, they dissaociate and also form aggregates.
- Aggregates can get bigger and bigger and form fibulary tangles.
- Theres 2 main camps of alzheimers research: tau and amyloid researchers. Many people look to target the tau component and advocate greately for it. Majority looking at amyloid.
- Amyloid: amyloid precursor fragment is embedded in the membrane, the fragment partially embedded in membrane
Lecture 11: Amyotrophic Lateral Sclerosis (ALS)
- Signs and symptoms of ALS
- Signs:
- Symptoms:
- Upper/lower motor neurons.
- Muscle strength, control, overall control of musculature.
- Neurodegeneration in ALS and systems affected
- Upper/lower neurons very spatially seperate
- motor cortex
- ventral horn of spinal cord
- either can start degenerating
- Upper/lower neurons very spatially seperate
- Selective vulnerability in neurodegenerative diseases
- Large fraction of motor cortex are BETS cell, very LARGE (20x larger than other non BETS cells)
- However lower motor cells are not as large. Does this eliminate it as candidate?
- Large fraction of motor cortex are BETS cell, very LARGE (20x larger than other non BETS cells)
- Relation of gain and loss of function to pattern of inheritance
- Studying the genetics of neurodegenerative diseases
- Not the genetics itself, but the STUDY of the genetics
- What do we have that lets us find answer?
- What are their limitations?
- Large genome studies
- Study variations in genomes, even at single gene levels
- Genetic mutations related to ALS
- SOD1 mutation: Multiple different ways to have mutations here
- Some are autosomal dominant
- Connect patterns of inheritance to patterns of gain/loss of function
- TDP43 and Fuss
- Studies showing that both gain and loss interact in complex ways to lead to increased risk
- C9ORF72: Mutation associated with both ALS and with frontal temporal dementia (FTD)
- Even though both are distinct, one mutation confers risk to both
- Risk factors affecting different systems
- IE affecting oxidative stress. Stress up, could more heavily affect Betz cells as a spitball
- SOD1 mutation: Multiple different ways to have mutations here
- Cell autonomous and non-cell autonomous mechanisms of toxicity
- Cell autonomous: Something in the cell alone that is leading to toxicity to the cell
- Non cell autonomous: Not that, not the cell alone. Other cells/Other cell types doing this
- Excitotoxitcity and relation to ALS
- Two excitatory neurons one connected to other(upper->lower) and excess glutamate released could lead to post synaptic toxicity through the mechanisms of excitotoxicity
- Potentially one player in ALS
- If a lower motor neuron degenerates first, and the upper neuron, and there is no lower->upper projection, could the lower neuron be responsible? No, lower neuron would not project,could not be responsible
- Two excitatory neurons one connected to other(upper->lower) and excess glutamate released could lead to post synaptic toxicity through the mechanisms of excitotoxicity
- Spreading of pathology in neurodegenerative diseases
- Protein folding and prion-like protein interactions
🧪 -> Refresh the Info
Did you generally find the overall content understandable or compelling or relevant or not, and why, or which aspects of the reading were most novel or challenging for you and which aspects were most familiar or straightforward?)
Did a specific aspect of the reading raise questions for you or relate to other ideas and findings you’ve encountered, or are there other related issues you wish had been covered?)
🔗 -> Links
Resources
- Put useful links here
Connections
- Link all related words